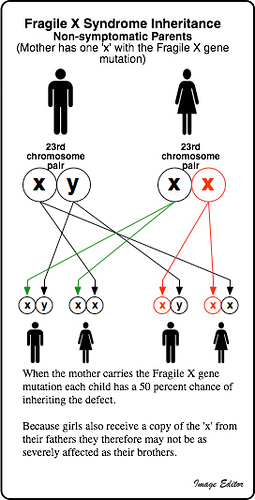
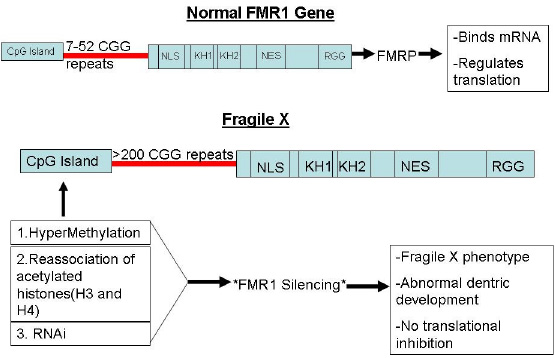
Sindrome X fragile (Sindrome di Martin Bell). La sindrome della X fragile costituisce la forma più frequente di ritardo mentale ereditario. Essa è causata da una mutazione del gene FMR1 localizzato sul cromosoma X (mutazione FRAXA). Questa mutazione consiste in un’amplificazione ed in una successiva metilazione di una sequenza di triplette CGG localizzata nella porzione trascritta e non tradotta del primo esone del gene ed è responsabile di un blocco della sua trascrizione. Poiché il gene codifica per una proteina necessaria al normale sviluppo del sistema nervoso centrale, la mancanza della proteina è causa di ritardo mentale. Gli alleli normali presentano un numero di triplette compreso tra 5 e 45; negli alleli mutati, unici responsabili della sindrome, questo numero è superiore a 200 (mutazione completa). Alleli con un numero di triplette compreso tra 56 e 200 (premutazione) sono normalmente espressi ma risultano instabili, con tendenza alla transizione verso la mutazione completa nel corso della meiosi femminile. Pertanto, donne sane che presentano una premutazione hanno un elevato rischio di trasmettere una mutazione completa ad un figlio o ad una figlia. Alleli con un numero di triplette compreso tra 46 e 55 rappresentano i cosiddetti alleli intermedi, alleli cioè considerati normali ma suscettibili di subire espansioni o regressioni con possibilità di diventare alleli premutati nelle successive generazioni. Si ritiene che la prevalenza delle donne portatrici di premutazione nella popolazione generale sia pari a circa 1:260. Il ritardo mentale correlato con mutazioni del gene FMR1 nei maschi affetti è solitamente di grado medio o grave, nelle donne affette è solitamente di grado lieve. Nei maschi affetti sono spesso presenti altri segni clinici come statura e circonferenza cranica superiori alla media, faccia allungata con mento prominente, padiglioni auricolari sporgenti, aumento del volume di testicoli (macroorchidismo) postpuberale, disturbi del comportamento e, meno frequentemente, epilessia, strabismo ed ipotonia con scoliosi. Le donne affette possono presentare solo in forma sfumata alcuni dei segni clinici descritti nei maschi. Peraltro, esistono anche maschi con ritardo mentale e con mutazione completa del gene FMR1 che non presentano i segni clinici caratteristici della sindrome. Non è, quindi, possibile escludere la mutazione del gene FMR1 in un maschio con ritardo mentale in assenza dei segni clinici caratteristici della sindrome della X fragile. Infine, la presenza di due cromosomi X nelle cellule femminili e la casuale e precoce inattivazione di uno solo di questi nelle cellule embrionali rendono ragione del fatto che una terzo circa delle donne portatrici di una mutazione completa manifesta ritardo mentale.
Indicazioni al test. L’analisi molecolare del gene FMR1 (analisi del DNA) è indicato sia in soggetti sintomatici che in soggetti sani. Nel primo caso il test assume valore diagnostico ed è indicato in soggetti di sesso maschile con ritardo mentale con segni clinici suggestivi della sindrome della X fragile o per i quali, comunque, non sia possibile porre diversa diagnosi del ritardo, ed in soggetti di sesso femminile con ritardo lieve per i quali non sia stata riconosciuta altra causa del disturbo. Poiché in donne portatrici della premutazione altrimenti normali si registra una menopausa precoce con frequenza significativamente più alta rispetto alla popolazione generale, la menopausa precoce costituisce un’ulteriore indicazione all’esame. Recentemente è stato osservato che donne portatrici della premutazione, sane fino all’età adulta, possono sviluppare tardivamente segni neurologici rappresentati da tremori ed atassia. Pertanto, il test molecolare è anche indicato in soggetti che presentano un quadro clinico di tal genere non riconducibile ad altra causa nota. Tra i soggetti sani di sesso femminile, particolarmente in presenza di volontà riproduttiva, l’esame molecolare è, invece, indicato per individuare le portatrici della premutazione, o, più raramente di una mutazione completa, in quanto esse costituiscono una categoria di donne ad alto rischio di procreare un figlio affetto. La probabilità di essere una portatrice sana aumenta rispetto alla media della popolazione femminile in caso di parentela con maschi affetti dalla sindrome della X fragile o da ritardo mentale non altrimenti diagnosticato. Solo nel caso in cui una donna è risultata essere portatrice sana di una premutazione o di una mutazione completa, è indicata la diagnosi prenatale sul DNA fetale. Il test prenatale sul feto è indicato solo dopo l’esecuzione di un test sul DNA materno che abbia dimostrato la presenza di una condizione di rischio aumentato per la sindrome della X fragile nel feto (presenza di premutazione o mutazione completa nella madre). Infine, nel caso in cui l’esame molecolare sul feto dimostri una mutazione completa, in caso di feto maschio non è possibile prevedere il grado di ritardo mentale che ne deriverà, in caso di feto femmina non è possibile predire se la mutazione determinerà oppure no ritardo mentale.
Analisi molecolare. La diagnosi molecolare per studiare la Sindrome dell’X-Fragile e i disordini ad essa correlati viene effettuata valutando lo stato di espansione delle ripetizioni CGG e (laddove indicato) di metilazione del gene FMR1. L’analisi viene condotta con un kit che utilizza il metodo della Repeat-Primed PCR (RP-PCR) (Adler K et 2011, Chen L et al 2010) che, consente di individuare mutazioni complete fino ad almeno 1300 ripetizioni CGG, di valutare accuratamente il numero di ripetizioni fino a 200 CGG, di individuare le interruzioni AGG (che rendono la ripetizione più stabile) e di risolvere in maniere inequivocabile la zigosità femminile. La sensibilità del test è superiore al 99% in quanto, sulla base delle attuali conoscenze, la quasi totalità delle mutazioni responsabili della sindrome della X-fragile e delle condizioni cliniche ad essa correlate è costituita dall’espansione del numero delle triplette CGG del primo esone del gene. In casi specifici, in presenza di mutazioni complete, è opportuno eseguire un approfondimento per valutare anche lo stato di metilazione dell’espansione.